
关键词:酮洛芬; 药剂学; 制备工艺; 缓释; 控释; 经皮给药;
Progress of Ketoprofen in Pharmaceutical Research
Abstract:Ketoprofen is an excellent non-steroidal anti-inflammatory drug, which is widely used in clinic, but with severe gastrointestinal adverse reactions. In recent years, many scholars have carried out numerous studies on pharmacy preparation technology to improve the bioavailability and reduce the toxic side effects of ketoprofen, such as packet complexes, ethosomes, sustained release, controlled release, etc…In this paper, the more advanced pharmaceutical preparation technology for ketoprofen in China is summarized for reference, so that more scholars can carry on further study, perfect or develop superior sustained-released, controlled released and transdermal drug formulations, and transform the laboratory technology into industrial production technology as soon as possible.
Keyword:Ketoprofen; Pharmaceutics; Preparation Technology; Sustained Release; Controlled Release; Transdermal Drug Delivery;
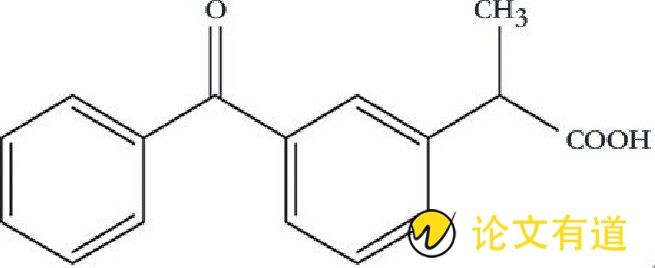
酮洛芬 (ketoprofen, KP) , 化学名称2- (3-苯甲酰基苯基) 丙酸, 亦称间苯甲酰氢化阿托酸, 其化学结构见图。其结构式见图1.
本品为白色结晶性粉末, 无臭或几乎无臭, 在甲醇中极易溶解, 在乙醇、丙酮或乙醚中易溶, 在水中几乎不溶, 是法国1976年研制的苯丙酸类非甾体抗炎药, 其主要作用机制为可逆性抑制环氧酶和脂氧酶, 从而抑制前列素和白三烯的生物合成, 发挥解热、镇痛和抗炎作用, 亦有中枢止痛作用。本品镇痛作用优于同类药物且维持时间长, 消炎作用较布洛芬强, 不良反应小, 毒性低, 安全性及耐受性好。临床上主要用于治疗风湿性或类风湿性关节炎、骨关节炎和强直性脊椎炎等。
1、KP的药代动力学研究
口服易自胃肠道吸收, 其吸收过程受食物影响, 口服生物利用度低且血药浓度个体差异大, 半衰期为1.6~1.9 h, 有效血药浓度维持时间短, 而连续服药对胃肠道有刺激作用, 长期服用会导致消化道溃疡、出血等症状[1].1次给药后, 约0.5~2 h可达血浆峰浓度, 每日3次, 每次200 mg, KP多剂量达稳态过程峰浓度范围为 (3.27±4.65) ?g/m L, 血药浓度KP有效治疗应该在浓度0.1?g/m L左右[2].在血中与血浆蛋白结合力极强, 在24 h内血尿中排出率为30%~90%, 其排出物大多为结合形式[3].静注单剂量KP符合二室模型, 可完全在肝脏内代谢, 该药即无酶诱导作用亦无抑制其自身代谢的作用, 其在关节滑囊中的浓度只有血浆浓度的1/3, 且持续时间较长[4].
2、KP现代给药系统的研究
目前, 国内外上市的剂型主要有片剂、胶囊剂、注射剂、栓剂、外用洗剂等。为了增加药物的溶解度, 减少不良反应和提高生物利用度, 近年来, 国内外学者对KP的新剂型及其制备工艺进行了深入研究, 尤其缓释制剂、经皮外用制剂 (可避免肝脏的首过作用) , 减少或避免对胃肠道的不良反应等。
2.1 新型固体分散体
固体分散体是将药物以分子、胶体、无定形或微晶状态分散在载体中, 以提高药物溶出度的体系。传统溶剂挥发法制备固体分散体有许多问题:过程复杂, 制备的固体分散体粉碎过筛压片;稳定性不好, 易老化;药物易结晶、聚结;干燥过程中共沉淀物黏度越来越高, 很难将有机溶剂快速有效去除。聂伟等[5]使用高压静电纺丝技术制备固体分散体, 以水难溶性药物KP为模型、聚乙烯吡咯烷酮 (PVP) K30为成纤基材, 将KP 15 g溶于无水乙醇100 m L中加入PVP-K30 30 g, 于常温、150 r/min搅拌2 h后, 超声脱气 (200 W, 15 min) , 得淡黄色的清亮纺丝液, 纺丝液流速2.0 m L/h, 接受板至喷口距离15 cm, 电压10 k V, 环境温度 (25±1) ℃, 相对湿度 (64±2) %, 制备的载药纤维毡, 然后根据需要进一步加工成所需的缓释给药系统, 制备得到的是纳米纤维状固体分散体具有连续三维立体网状结构的特征, 表面积巨大, 孔隙率高, 比较原药粉末、PVP-K30粉末、纳米纤维状固体分散体, X-射线衍射和差示扫描热分析结果表明, 药物以无定形或分子状态高度分散于聚合物基材中;相同比较, 傅里叶红外光谱分析结果证明, KP与PVP之间能通过氢键相互作用。实验显示溶于水的速度快, 体外溶出结果:载药纤维毡中的KP能在30 s内释放完全, 是原药粉溶出速率的30倍, 明显优于传统的固体分散体, 但该工艺的缺陷是目前还不能工业化生产。
2.2 微球、微丸
乳化溶剂扩散法是将药物与高分子载体材料溶解在良溶剂或良溶剂与架桥剂的混合溶剂中, 在搅拌条件下加入到溶有乳化剂且对药物和高分子载体材料为不良溶剂的连续相中制成O/W型乳滴, 然后伴随良溶剂的迅速扩散至不良溶剂中, 药物和载体材料共同析出成球形颗粒。王岩等[6]就是采用该方法制备KP微球, 将处方量的KP超声溶解在定量的乙酸乙酯中, 将其缓慢滴加到含有处方量十二烷基硫酸钠的饱和乙酸乙酯的水溶液中制得乳状液, 继续搅拌乳化10 min, 加入过量水, 有机溶剂不断扩散至水中, 乙基纤维素和KP的溶解度降低、共沉在乳滴中形成微球, 静置后抽滤、水洗、干燥即得。该处方的优化是依据微球的收率、包封率为考察指标, 考察投料质量比、乙基纤维素的质量浓度、油水相体积比、乳化剂用量等4因素, 采用L9 (34) 正交试验设计, 得到最佳处方分别是:1∶1、80 g/L、1∶5、15 g/L, 制得微球粒径分布在20~80?m内, 平均粒径49.78?m, 收率73.3%, 包封率91.0%, 微球体外释放测定相比原料药具有明显的缓释特性, 该制备工艺简单可行, 采用该工艺还可制备肠溶微球[7], 胃漂浮微球[8]等。
李海刚等[9]制备KP微丸缓释片剂, 首先将KP与微晶纤维素、乳糖制备软材, 采用挤出-滚圆法制备KP素丸, 干燥, 过筛, 取20~30目药筛之微丸备用, 用Eudragit®RS30D和Eudragit®RL30D包衣, 以释放度为考察指标, 考察Eudragit®RS30D和Eudragit®RL30D的用量、比例、包衣液中聚合物含量、包衣过程中的干燥温度、时间、蠕动泵流速、喷雾压力、刀盘转速, 分别是20%、比例1∶0.2、10%、60℃、12 h、2.0 m L/min、0.6 MPa、300 r/min.取KP包衣丸与处方量的KP、乳糖、硬脂酸镁、微粉硅胶、羟丙基甲基纤维素 (HPMC) K4M等进行压片, 制得的片剂, 体外释放度测定:在2 h内能释药30%, 剩余70%药物在随后的10 h内缓慢释放。该缓释片工艺简单, 易于操作。
2.3 微乳
微乳是水、油、表面活性剂和助表面活性剂按适当比例混合, 自发形成的各向同性, 透明, 热力学稳定的分散体系。袁悦等[1]制备KP微乳, 首先测定KP在不同油、表面活性剂、助表面活性剂的溶解度, 确定药物的投料量为1.25%, 以伪三元相图为基础, 依据微乳区域大小, 初步筛选微乳处方, 以体外累积渗透量和稳态渗透率为指标, 以单因素考察, 进一步筛选微乳处方分别确定油相用量、混合表面活性剂比例及用量, 结果选用KP溶解度较好的肉豆蔻酸异丙酯为油相 (IPM) 3%, 混合表面活性剂 (Labrasol®24%/Plurol Oleique®6%) 30%和水, 含KP1.25%, 所制备的微乳 (O/W) 具有很好的稳态渗透速率: (46.47±2.1) ?g/ (cm2·h) (r=0.992 4) ;熊小英等[10]制备KP微乳凝胶, 也是以伪三元相图为基础确定表面活性剂Cre EL35与助表面活性剂Labrasol质量比为Km=1, 然后固定薄荷油和KP用量, 设计一系列处方, 将KP溶于处方中的表面活性剂、助表面活性剂和油相组成的混合液中, 25℃恒温水浴至药物溶解完全, 缓慢加入处方量的水, 搅拌至澄清透明O/W型KP微乳, 以累积渗透量和皮肤滞留量为指标, 用改进的Franz扩散池考察微乳处方各成分的用量最终确定最佳处方, 以黄原胶为凝胶基质, 将其置于水中充分溶胀, 逐步加入最佳处方制备的KP微乳中混合均匀, 静置脱气得KP微乳凝胶, 进行离体鼠皮透皮实验, 结果:KP微乳、KP微乳凝胶8 h的皮肤滞留量分别为 (705.13±33.18) ?g/cm2和 (502.75±28.54) ?g/cm2, 为对照组 (KP 30%乙醇溶液) 的4.66倍和3.22倍, 结果显示, 微乳作用后的皮肤角质层微观结构改变, 细胞间隙增加, 有利于药物的经皮吸收与局部滞留。微乳制剂有望成为KP新型经皮给药制剂。
2.4 β-环糊精及其衍生物包合物
环状糊精 (β-CD) 它可以包络各种化合物分子, 增加被包络物对光热、氧的稳定性, 改变被包络物质的理化性质。本品可与多种化合物形成包结复合物, 使其稳定、增溶、缓释、乳化、抗氧化、抗分解、保温、防潮, 并具有掩蔽异味等作用, 为新型分子包裹材料, 其衍生物磺丁基-β-环糊精 (SBE-β-CD) 的水溶性远高于β-CD, 可提高难溶性药物的水溶性, 服后便于体内吸收, 提高生物利用率。韩瑞亭[11]利用β-CD制备KP包合物, 通过正交试验法优选投料比、温度和时间分别是主客摩尔比1∶2、于25℃电动搅拌3 h:取KP 0.5 g, 95%乙醇100 m L置于三角瓶中, 35℃水浴搅拌溶解, 放冷至25℃, 搅拌加入β-CD 4.4 g使之溶解后, 电动搅拌2.5 h后, 搅拌加入氯仿15 m L, 继续搅拌0.5 h, 得混悬液, 置冰箱冷藏24 h, 过滤、乙醇洗2次、干燥、粉碎、过筛 (60目) , 备用。其制备方法简单, 制得的包合物包合率和收率较高。包合物相比KP药物溶出率测定数据、水解稳定性实验数据、动物口服实验数据显示分别是:包合物溶出率高、包合物能增加药物的稳定性、包合物的相对生物利用度高, 完全可以制备颗粒剂后降低药物的刺激性和毒副作用。归银兰等[12]使用水溶性更好的SBE-β-CD制备KP包合物, 采用冷冻干燥法, 依据平衡相溶解度实验数据得到主客分子摩尔比1∶1, 换算成重量比约1∶9 (w/w) , 取KP 2.56 g加乙醇5 m L室温溶解, 滴加入35 m L SBE-β-CD的饱和水溶液中, 边加边搅拌, 继续搅拌2 h, 静置12 h后, 将混悬液置于冷冻真空干燥机中真空干燥, 即得KP包合物24.98g.比较SBE-β-CD、KP、物理混合物、包合物, 差示扫描热分析仪扫描图谱验证KP被SBE-β-CD包合, 取KP包合物20 mg分别测定包合率及KP的含量分别是95.14%, 9.98%, 数据说明包合率高, 在包合过程中KP损失很少, 绝大部分药物被包合;饱和溶解度实验显示包合物相比KP溶解度增加480倍;溶出实验显示KP-SBE-β-CD包合物30 min的溶出度可达90%, 而KP只有35%;利用8只健康SD大鼠分两组分别灌胃给药KP和KP-SBE-β-CD包合物, 测得数据显示KP-SBE-β-CD包合物的吸收比KP快, 血药浓度高, AUC是KP的2.8倍。以上制备工艺说明β-环糊精、SBE-β-CD的包合物能增加KP的溶出度及提高生物利用度, 制备工艺和方法简单, 可再进行剂型转换如颗粒剂、胶囊剂等。
2.5 固体脂质纳米粒
固体脂质纳米粒 (SLN) 是指粒径在10~1 000 nm, 以毒性低、生物相容性好、生物可降解的固态天然或合成的类脂为载体, 将药物吸附或包裹于脂质膜中制成的新一代纳米粒给药系统。李楠等[13]采用微乳法, 将KP和硬脂酸在65℃水浴上融化, 加入适量相同温度的Km=1∶4的吐温-80-乙醇溶液及蒸馏水, 于涡旋混合仪上涡旋1 min后, 即形成透明O/W微乳, 使用微乳注入装置距离2℃冷水分散介质液面6 cm, 在电磁搅拌下 (1 000 r/min) 将以1滴/5 s的速度滴入, 当全部注入后继续2℃保温搅拌15 min, 将微乳微热至4℃即得固体脂质纳米粒。首先以上方法重复制备空白微乳, 通过伪三元相图筛选出空白微乳优化处方:65℃将吐温-80与助乳化剂无水乙醇按Km=1∶4混匀, 再按10∶1的比例与硬脂酸混匀, 滴加蒸馏水后涡旋1 min保温, 直至透明微乳形成, 在4℃和室温条件下分别存放6个月, 期间观察澄清透明, 稳定性好, 以粒径为考察指标, 分别以不同乳化剂、不同助乳化剂、不同Km和相同油含量的空白微乳初步确定处方组成:Km=1∶4的吐温-80-乙醇溶液。依据之前预实验单因素考察的结果, 确定KP-固体脂质纳米粒 (SLN) 制备工艺参数影响显着性依次为:硬脂酸用量 (A) 、药物用量 (B) 、冷水温度 (C) 和微乳保温温 (D) , 分别对粒径、包封率及载药量测定, 采用L9 (34) 正交试验优化处方:A (0.5g) >C (2℃) >D (65℃) >B (25.0 mg) .依据最优处方制备KP-SLN的平均粒径 (143.9) , 包封率81.47%, 载药量8.16%.本方法稳定可靠, 具有增效减毒、缓控释作用。
2.6 二元醇质体
二元醇质体是醇质体的改进, 特殊的脂质体载体, 是按一定体积比乙醇与丙二醇的混合液制备二元醇质体, 近年成为透皮吸收药物的研究热点, 其透皮速率及皮肤滞留药量更大, 成品稳定性更好, 易于保存, 明显优于醇质体和脂质体。其透皮增强的主要原因, 一方面, 醇尤其丙二醇本身就具有促渗透作用, 可增加药物在角质层的溶解度, 改变角质层脂质的相变温度, 增加其流动性、柔韧性;另一方面, 醇混合液的加入使醇质体的粒径更小, 囊泡结构的流动性更强, zeta电位也发生了利于渗透的变化。王军等[14]采用乙醇注入法, 称取处方量的KP和卵磷脂, 用无水乙醇/丙二醇混合液溶解, 在封闭条件下搅拌, 同时细流加入磷酸盐缓冲液 (PBS) , 加完后继续搅拌30 min, 温度30℃, 再于冰浴下探头式超声30 s, 即得二元醇质体, 其凝胶剂采用研合法即得。以包封率为考察指标, 依据预实验结果, 固定卵磷脂和KP的质量比为9∶1, PBS的p H值为7.0, 醇的体积分数为30%, 优选乙醇与丙二醇较佳的比例为7∶3.在以上条件下制备的二元醇质体其平均包封率为73.85%, 其平均粒径为289.86 nm.相同条件下制备的醇质体凝胶和二元醇质体凝胶体外离体鼠皮渗透试验比较, 二元醇质体凝胶累积渗透量 (Q) 为醇质体的1.8倍, 稳态透皮速率 (J) , 皮肤药物滞留量 (RA) 也显着增加, 结合凝胶技术二元醇质体分布于凝胶三维网状结构中, 其稳定性更好, 完全可以开发外用经皮吸收制剂。
2.7 电纺纤维膜
药物与高分子聚合物通常溶解于有机溶剂中制成纺丝液, 混合纺丝液被注射泵挤压至针头处并形成小液滴, 在高压电场作用下形成超细纤维纳米或亚微米级的聚合物纤维膜加工技术, 可实现药物的缓控制释放和靶向给药。
将KP与PVP溶于无水乙醇制成混合纺丝液, 通过静电纺丝技术制成纤维膜, 体外溶出结果表明, 所制备的纳米纤维状固体分散体中KP能在30 s内释放完全[15], 可以用作药物速释材料模型;将KP与乙基纤维素通过静电纺丝技术制成载药纳米纤维膜, 释药结果表明, KP在7 d左右的累积释药率达到50%左右, 可以用作药物缓释材料模型[16].
2.8 透皮贴剂
透皮镇痛贴剂是较为成熟的一类透皮贴剂, 应用广泛, 代表药物主要有芬太尼和KP透皮贴剂, KP有较好的水溶性, 适合做成透皮制剂, 主要用于治疗骨关节炎, 已知的KP透皮制剂有贴剂、凝胶剂、乳剂、软膏以及基于离子导入技术的贴剂。
将KP原料药及处方量的乙酸乙酯以及促渗剂 (肉豆蔻酸异丙酯) 溶解于压敏胶 (丙烯酸酯) 中搅拌均匀, 涂布制备压敏胶分散型贴剂, 体外经皮渗透实验结果表明, 以肉豆蔻酸异丙酯质量分数2.5%作为促渗剂制备贴剂, 其经皮渗透性与参比贴 (Mohrus®) 一致[17].
3、结语
KP作为优良的非甾体抗炎药, 上述缓释控释制备工艺以及经皮给药工艺主要停留在实验室阶段, 在增效减毒以及提高生物利用度方面效果显着, 但相应的实验研究文献不是很多, 本人进行归纳整理, 供研究者参考, 便于学者进一步研究完善上述缓释控释制备工艺以及经皮给药工艺或开发出更好的缓释控释及经皮给药制剂, 把实验室技术尽早转化为工业生产技术, 尽早用到临床造福患者。
参考文献
[1]袁悦, 李三鸣, 王思玲, 等。酮洛芬微乳经皮给药制剂的研究[J].中国药学杂志, 2006, 41 (21) :1647-1650.
[2]刘新宇, 吴芳, 陈连剑。高效液相色谱法测定酮洛芬的血药浓度[J].中国药科大学学报, 2001, 32 (4) :279-281.
[3]陈琰, 胡晋红, 范国荣, 等。酮洛芬的制剂研究[J].药学进展, 2001, 25 (4) :219-223.
[4]徐美英。止痛新药-酮洛芬[J].《国外医学》麻醉学与复苏分册。1992, 13 (6) :325-327.
[5]聂伟, 余灯广, 申夏夏, 等。电纺制备酮洛芬纳米纤维状固体分散体[J].中国医药工业杂志, 2010, 41 (5) :344-347.
[6]王岩, 冀艳艳, 徐璐, 等。乳化溶剂扩散法制备酮洛芬缓释微球[J].中国药剂学杂志 (网络版) , 2012, 10 (4) :62-66.
[7]冀艳艳, 王中彦, 王岩, 等。乳化溶剂扩散法制备酮洛芬肠溶微球[J].中国药剂学杂志 (网络版) , 2013, 11 (3) :45-50.
[8]冀艳艳, 陈磊, 王中彦, 等。酮洛芬漂浮微球的制备及体外释放[J].中国药剂学杂志 (网络版) , 2012, 10 (5) :79-85.
[9]李海刚, 袁中文, 管世侠, 等。酮洛芬微丸缓释片的制备及体外释放度考察[J].中国医院药学杂志, 2011, 31 (20) :1710-1713.
[10]熊小英, 李娟。酮洛芬微乳凝胶的制备及透皮机制[J].中国药科大学学报, 2012, 43 (6) :514-518.
[11]韩瑞亭。酮洛芬β-环糊精包合物制备及质量评价[J].黑龙江医药, 2011, 24 (2) :228-229.
[12]归银兰, 归小龙。酮洛芬磺丁基醚-β-环糊精包合物的制备[J].中国医院药学杂志, 2012, 32 (20) :1621-1625.
[13]李楠, 孔令钰, 刘佩莉, 等。酮洛芬固体脂质纳米粒的制备[J].天津药学, 2014, 26 (6) :18-22.
[14]王军, 何文。酮洛芬二元醇质体凝胶的研制及其质量考察[J].中国药师, 2012, 15 (6) :780-782.
[15]聂伟, 余灯广, 申夏夏, 等。电纺制备酮洛芬纳米纤维状固体分散体[J].中国医药工业杂志, 2010, 41 (5) :344-347.
[16]刘凯琳, 吕瑶, 桑青青, 等。静电纺丝制备乙基纤维素纳米载药纤维[J].纺织导报, 2016, 25 (12) :52-55.
[17]李龙伟, 孙英华, 王金兰, 等。酮洛芬贴剂的体外经皮渗透性考察[J].中国药剂学杂志 (网络版) , 2016, 14 (1) :11-17.